登录
记住用户名密码
记住用户名密码
数据驱动机器学习研究方法可以揭示催化构效关系等复杂规律,进而对材料的关键性质进行快速预测,从而可以极大地加速新材料的筛选和合理设计。但实际研究中,面对庞大的待研究材料空间,构建描述大材料空间数据特性的机器学习模型要求的数据量往往超出大多数实验室的实验或者计算能力。特别是对于催化材料研究,催化活性和选择性的耦合往往导致了二者不可兼得的“跷跷板”效应。因此开发面向基于实际小样本搜索大材料空间的多目标约束的材料设计框架对于新型催化剂开发和设计尤为关键。
针对上述问题,华中科技大学微纳中心单斌教授团队与郑州大学李庆奎教授先进靶材料研究中心团队在化工催化领域知名期刊《Chemical Engineering Journal》上发表了最新研究成果,论文标题为“Active learning driven discovery of novel alloyed catalysts for selective ammonia oxidation(主动学习驱动的氨气选择性氧化合金催化剂发现)”,DOI: https://doi.org/10.1016/j.cej.2024.152300。郑州大学先进靶材料中心青年教师杨家强为论文第一作者,单斌教授和李庆奎教授为本文的共同通讯作者。
本研究提出了一种基于贝叶斯优化的多目标约束的新型合金催化剂主动学习搜索策略,旨在为氨气选择性氧化 (NH3-SCO) 过程等复杂催化过程寻找兼具反应活性和选择性的催化体系,如图1所示。

首先基于过渡金属元素构建了包含超过1000种不同表面局域构型的合金体系大空间,主要是Ag、Ag、Cu、Pd、Pt、Ir 和Rh七种金属作为基体元素,过渡族金属中除去有毒有害元素之外元素作为掺杂元素构建低指数晶面上的表面局域构型,如图2所示。另一方面,采用过去研究中得到~300种Ag、Ag、Cu、Pd、Pt、Ir 和Rh七种金属互相掺杂得到的合金表面局域构型数据作为小样本数据[J. Mater. Chem. A, 2022,10, 25238-25248]。基于小样本数据集训练面向NH3-SCO反应活性与选择性描述符的高斯过程回归模型(GPR)。过去研究发现NH3-SCO金属催化剂上当N结合能E(N)~4.50 eV时催化活性最优,同时氧原子E(O)结合越强N2选择性越高[J. Mater. Chem. A, 2022,10, 12447-12457]。

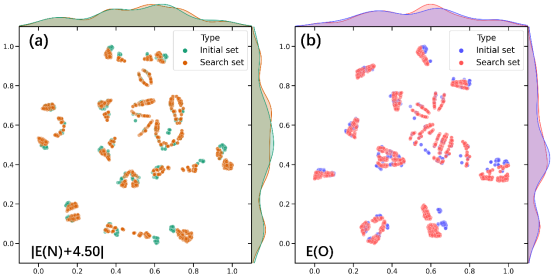
进一步将纯相体系表面E(N) 和E(O)作为先验知识特征,表面和次表面原子平均信息以及晶面特征信息作为结构特征,共同构建了面向催化活性和选择性描述符预测的特征空间。注意,为了降低描述符之间的相关性,对活性描述符转化为|E(N)+4.50|,这样可以将求最接近中间值转变为求最小值。基于以上特征的小样本和大空间的数据分布如图3所示。可以发现,两者的特征空间分布存在明显差异,这意味着基于小样本的机器学习模型可能并不能适用于大空间的材料发现。
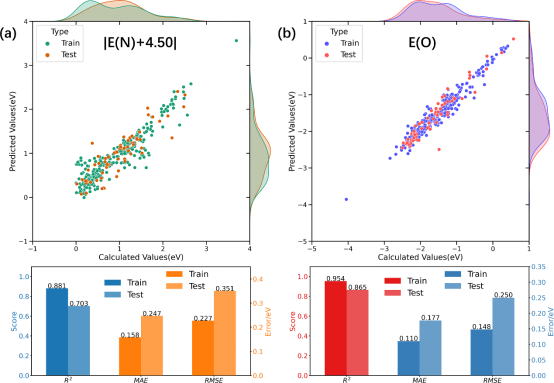
基于以上特征和描述符数据,进一步构建了催化活性描述符|E(N)+4.50|和选择性描述符E(O)预测的高斯过程回归模型(GPR),如图4所示。可以发现GPR模型展示了较好的预测精度和泛化能力,特别是E(O)预测模型在测试集的MAE低于0.177eV,R2~0.865。
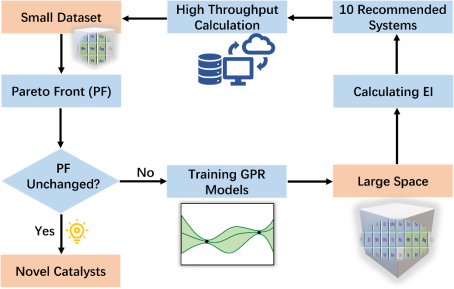
基于得到的小样本高斯过程回归预测模型,结合帕累托解和贝叶斯优化方法建立多目标约束的合金催化材料理性设计的主动学习框架,如图5所示。该框架通过帕累托解策略处理多目标任务,选择数据集中非支配解作为兼顾活性和选择性的候选体系;同时可实现初期侧重于对未知区域的采样,提升描述符数据预测模型的精度,在后期侧重于对已知区域内潜在体系的搜索,兼顾探索-利用。具体流程如下:1) 采用NH3-SCO催化描述符预测的小样本机器学习模型计算整个大材料空间体系的描述符数值和不确定度,并计算对应体系的采集函数EI函数数值。2) 选择EI数值最大的 10个体系并高通量计算对应的真实描述符数据。3) 预处理新体系的特征和描述符数据,加入并更新原始数据集,确定新数据集的双目标的帕累托最优解和前沿。4) 基于新数据集重新训练描述符预测模型。5)迭代以上步骤,直至相邻两次迭代的帕累托前沿不再变化,便可终止迭代。
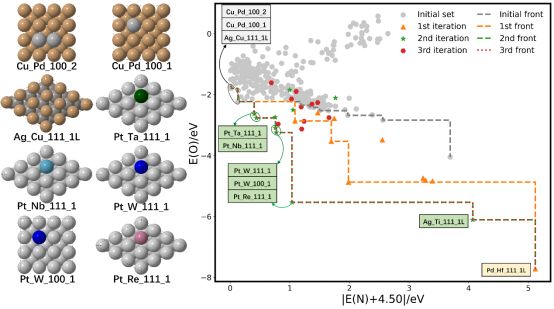
基于以上框架,我们发现经过三次迭代共计30个构型的高通量计算,NH3-SCO合金催化剂主动学习搜索即收敛,如图7所示。进一步对位于前沿和近邻前沿的合金体系进行简化反应机理的高通量计算,发现帕累托前沿上10种候选体系中的6种体系,7种前沿近邻体系中的3种,共计9种合金体系是潜在的兼顾活性和N2选择性的催化体系。进一步合金局域结构稳定性理论计算也表明这些体系在热力学上是稳定的。
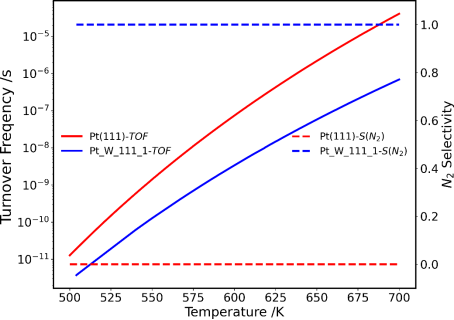
进一步考虑完整NH3-SCO催化反应过程和微反应动力学方法研究了不同反应温度下催化性能,如图7所示。选择Pt_W_111_1候选体系作为代表,与Pt(111)晶面的催化性能进行对比,可以发现,Pt_W_111_1尽管展示出略低于纯相体系的本征催化活性(TOF),但具有远超于纯相体系的N2选择性,实现了催化活性和选择性的合适取舍,因此更具有实际应用价值。这也进一步验证了主动学习搜索结果的合理性。
总结
本研究基于来自过去研究中的小样本数据训练催化描述符预测模型,并结合帕累托解策略处理催化活性和选择性双目标约束,采用基于贝叶斯优化的主动学习实现大空间最优值搜索,实现了小样本搜索大空间,低成本高效率发现新型催化体系。本研究也证明了即使基于粗略的代理模型和稀疏不平衡局域小样本,主动学习搜索仍可以发现隐藏在庞大数据空间中的潜在材料体系。