登录
记住用户名密码
记住用户名密码
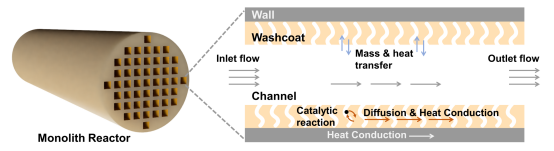
Monolith反应器(整体式反应器)被广泛用于尾气后处理、催化燃烧等过程,是气相催化领域的关键优化对象。一个可靠、细致的仿真模型,能够准确预测工况下反应器的转化效果,完整地描述催化过程的各种细节,进而为反应器、排放 系统的设计提供思路与引导。然而monolith反应器中的物理与化学过程涉及多个时间与空间尺度,以及多个物理场与表面化学反应的耦合,这给monolith的精确且高效建模带来挑战。
微反应动力学方法是一种基于第一性原理的表面催化动力学理论,其能够覆盖非常宽的温度和物种浓度变化范围,适用于复杂工况下的催化反应动力学计算。以微反应动力学作为表面反应求解器的一维模型是近年来monolith模拟领域的重要发展方向,但该方法的引入也给整体的计算效率和稳定性带来难题。机器学习是一种基于数据与“经验”的研究方法,能够实现高维度数据的分析处理,学习归纳科学问题中隐藏模式与规律,近年来被广泛用于材料模拟领域。这种数据驱动的方法有潜力解除计算效率的桎梏,辅助实现monolith仿真模型和微反应动力学模型的耦合,是构建新型monolith跨尺度模拟框架的关键。
华中科技大学单斌教授团队提出了一种结合monolith一维两相模型、微反应动力学和机器学习方法的跨尺度模拟框架:引入自主设计的速率变换函数,克服了因特殊的数据分布特征导致的机器学习模型不可用的难题;对比了支持向量回归、随机森林回归和极限树回归三种算法的效果,进一步将最优的极限树回归速率预测模型耦合到monolith模型中,最终实现工况下NH3-SCO催化体系反应器输出信息的精准、高效预测。相比与传统方法,基于极限树回归的模型在保持高准确性的基础上,计算时间降低50%以上,且在特定场景的稳定性上展现明显优势。
相关论文以“Extra Trees Regression Assisted 1D Monolith Reactor Simulations Based on Microkinetic Analysis and Rate Transformation”发表在Chemical Engineering Science上(DOI:10.1016/j.ces.2024.120721,https://www.sciencedirect.com/science/article/abs/pii/S0009250924010212),课题组硕士研究生陈荣昕、博士后杨家强(现任郑州大学青年教师)为共同第一作者,单斌教授、Chaitanya S. Sampara博士为共同通讯作者,华中科技大学为第一完成单位。工作得到了国家级稀土研究院李伟研究员的大力支持。

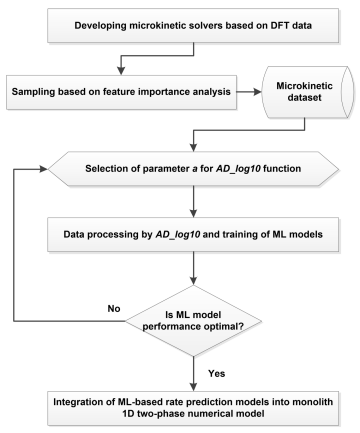
如图二所示,本工作建立了一个机器学习辅助的耦合微反应动力学与monolith一维模型的跨尺度工作流:首先基于密度泛函理论(DFT)计算得到的热力学与动力学信息建立平均场微反应动力学求解器,并在此基础上通过特征重要性分析确定采样策略,构建包含输入信息和基元步速率结果的预计算数据集。进一步采用自主设计的可调节双区对数前处理函数(AD_log10)调整数据分布,以克服训练机器学习模型时速率数据特异性带来的问题。最后,将速率预测模型集作为催化反应求解器,整合到催化器一维两相降阶模型中,实现从第一性原理计算结果到反应器实际催化效果的跨尺度评估。
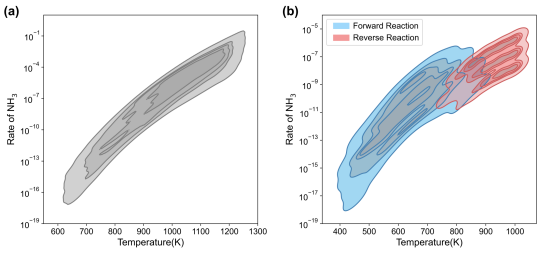
本工作选用Cu(100)和Cu(111)晶面上NH3选择性催化氧化反应作为探针反应验证跨尺度框架的实际效果,反应机理与第一性原理计算结果均来自于课题组之前已发表的工作,其中Cu(100)体系N2选择性好但活性较差,Cu(111)体系N2选择性较差但活性好,且随着环境变化关键基元步方向会发生改变。结合先验知识以及特征重要性分析,确定了两个体系温度及4个关键含氮物种的取值范围和采样数量,经微反应动力学计算构建数据集。图三展示了两个数据集以温度和NH3消耗速率为自变量的核密度估计结果,可以看出Cu(100)体系反应行为相对简单,R(NH3)与反应温度呈正相关;Cu(111)体系则相对复杂,尤其在700K至900K的温度范围内,数据集中正向和逆向反应同时存在,这中符号的变化也给后续机器学习回归模型的训练带来挑战。
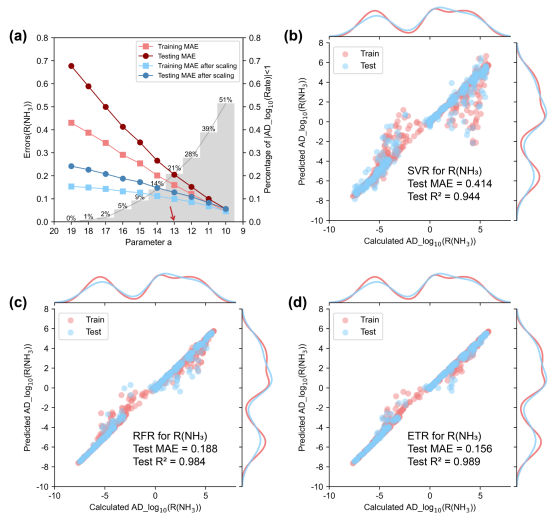
针对这个问题,我们创新性地提出了一种可调节双区对数前处理函数(AD_log10)对速率数据进行映射,其具体形式如图四所示。该函数是一个连续的分段函数,其包含一个可调整的参数a。图四 (a)展示了a=15时的函数形式:10ax>1或10ax<-1的区域称之为“激活区域”,映射的函数值几乎随x的10次方线性变化,效果接近于对数函数;-1<10ax<1时,即“抑制区域”,映射的函数值接近于0。总体上,该函数保留了对数函数级别的数据缩放功能,同时具备奇函数性质,能够适用于微反应动力学数据集中反应速率的前处理。图四 (b)展示了R(NH3)经a取不同值的AD_log10函数映射后的核密度分布结果,a越小则有越多的数据进入到“抑制区域”,其是机器学习训练过程中的一个关键优化参数。AD_log10函数是本工作中从微反应动力学到机器学习之间的关键桥梁,为整个跨尺度模拟框架提供了强力的支持。

基于已建立好的微反应动力学数据集,对比了支持向量回归、随机森林回归、极限树回归三种监督学习算法的效果,确定了模型的超参数以及a的最佳取值。首先,对于Cu(100)体系NH3-SCO反应,数据集中的目标量即各目标基元步的方向均不发生变化。因此,参数a的值仅取决于目标变量的最小值,即实际处理时将所有速率数据映射到函数的激活区。图五展示了监督学习算法对四个目标量在测试集上的平均绝对误差对比,可以看出三种模型均表现出不错的效果(且不存在过拟合现象),在测试集上R2值均超过0.99,MAE值均< 0.09且测试RMSE值均< 0.12。
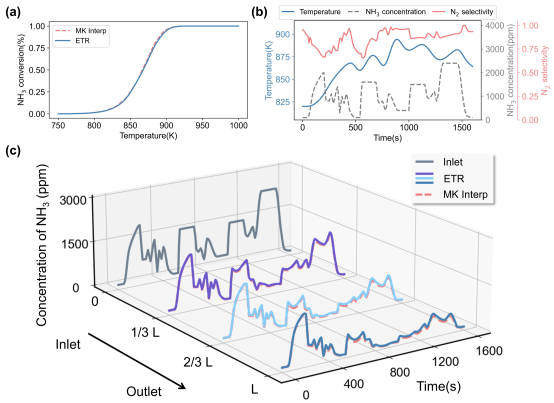
以训练好的ETR模型替代微反应动力学方法用于monolith一维两相模型中的表面反应计算。其中monolith一维模型的求解器基于MATLAB的ode15s函数构建,物理参数与模拟参数均根据实际应用确定。为了验证跨尺度模型的模拟效果,我们使用分段三阶Hermite插值多项式(PCHIP)方法基于原始微反应动力学数据集构建插值模型作为参照(传统方法)。图六展示了Cu(100)体系匀速升温和实际车载工况两种典型工况下的模拟效果,对于这个相对简单的体系,基于ETR的模型和基于微反应动力学插值的模型均表现出较好的响应性,两者预测结果差异较小,且基于ETR的模型分别降低了85%和55%的计算时间。
总结展望:
本工作提出了一种数据驱动的跨尺度模拟框架,将微反应动力学高效集成到monolith反应器模拟中。我们结合自主设计的可调节双区对数前处理函数,克服了微反应动力学计算的速率数据的特异性问题,训练出高精度的速率预测模型,并进一步耦合到monolith一维数值模型中。我们选取Cu(100) 和 Cu(111) 面上的 NH3 选择性催化氧化反应作为探针反应,以业界常用的基于插值的模型作为参比对象,以两种典型工况作为输入验证了模型的效果。与基于插值的模型相比,本工作提出的模拟框架在保持计算精度的基础上,在计算效率、稳定性等方面均展现显著优势。总体上,本工作提出的数据驱动的框架在耦合详尽微反应动力学分析的monolith车载仿真方面展现巨大潜力,也为化学反应工程领域中的跨尺度模拟提供了新的思路。