登录
记住用户名密码
记住用户名密码
以清洁能源H2为燃料的质子交换膜燃料电池应用前景广阔,而经蒸汽重整、水煤气变换等制备H2残留的微量CO将严重毒化Pt电极。富氢氛围中CO优先氧化(PROX)是进气路上在线去除痕量CO的理想方式,氧化铈等金属氧化物负载高分散过渡金属体系被认为是极具前景的PROX催化剂。
此前,华中科技大学单斌教授团队(微纳材料设计与制造研究中心)基于选择性ALD方法制备的Pt1/CoOx、Pt-Cu/CeOx等精细纳米结构表现出优异的CO氧化活性和选择性(J. Mater. Chem. A, 2020,8, 10180; Nat. Commun., 2020, 11, 4240),DFT计算结合微反应动力学分析阐明了CO与H2竞争氧化机制(ACS Appl. Mater. Interfaces, 2022, 14, 27762)。与单原子催化剂(SAC)相比,双原子催化剂(DAC)不仅具备高原子利用率和高选择性等优势,同时有望通过协同效应和电子调控效应等打破线性比例关系极限,实现PROX性能的进一步提升。对此,单斌教授团队与云南省贵金属新材料集团有限公司张爱敏首席研究员团队合作,在ACS Catalysis期刊发表题为“Synergistic Dual-Atom Catalysts on Ceria for Enhanced CO Preferential Oxidation: Insights from High-Throughput First- Principles Microkinetics”的研究文章。

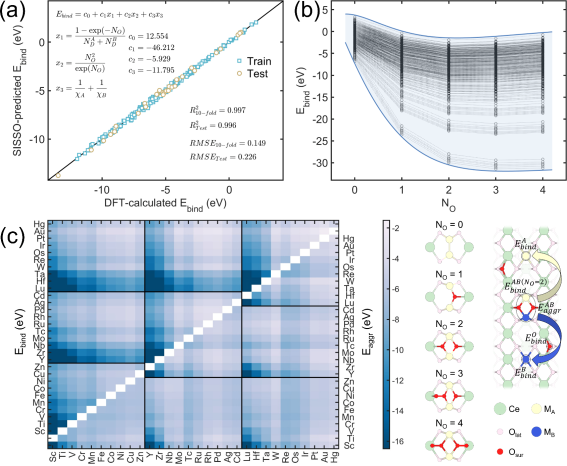
该项工作建立了包含了双原子模型构建、稳定性评估、反应网络DFT计算、微反应动力学分析和描述符识别等模块的高通量工作流。首先,以CeO2(110)为载体构建了包含3d, 4d, 5d过渡金属和5种氧配位环境组合的2325种负载双原子构型,如图1所示。进而通过不同机器学习方法建立稳定性预测模型,其中基于压缩感知的符号回归方法(Sure Independence Screening and Sparsifying Operator, SISSO)表现出良好的预测效果,如图2所示。SISSO推荐的3D描述符表明,双金属原子的D电子数之和、电负性倒数之和以及氧配位数是影响稳定性的关键,且D电子数更少、电负性更低以及氧配位数适中对应更稳定的DACs。
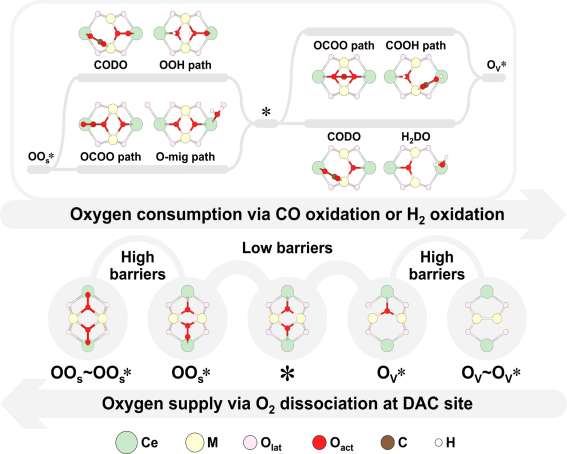
进而面向所有同质DACs(MA=MB),如图3所示,高通量计算CO直接氧化、H2直接氧化、OCOO、COOH、氧迁移和H(OH)辅助等可能发生路径中的关键基元步骤。结果表明,以适中氧配位数(NO=2,*)为原点的包含OCOO、O迁移和直接氧化等路径构成的MvK催化循环被认为是主导机制,即O2预解离的MvK机理。为验证该机理并量化分析影响CO氧化活性和CO2选择性的关键因素,以实验报道的Ir1-Ir1 DAC作为代表,基于CO PROX全反应网络的DFT计算能量信息,在实验条件下进行微反应动力学分析。
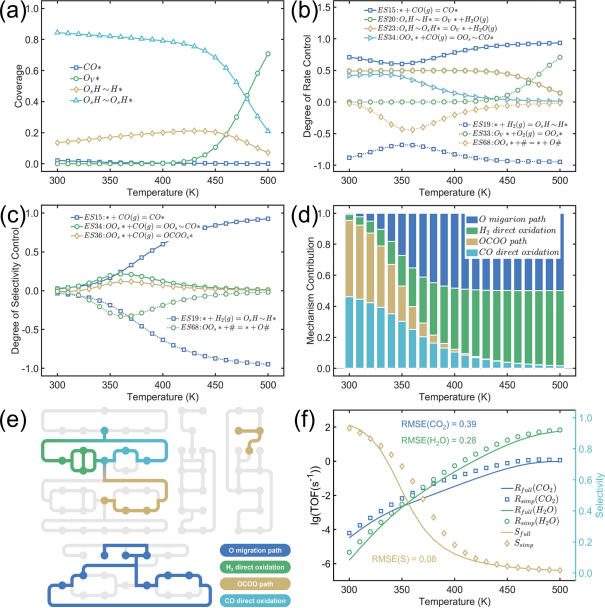
为实现对DACs PROX性能的整体评估,基于团队提出的“态到态”方法建立了复合微反应动力学模型。受负载双原子影响而显著区别于干净CeO2表面位点的区域被视为双原子限域位点,并对该区域进行整体化处理,即将限域位点中所有局域环境不同的可能共吸附组合视为相互独立的反应中间态,将基元反应视为相邻反应态之间的跳转。其中,氧迁移路径通过含氧物种的扩散沟通了双原子限域位点和干净CeO2表面位点(平均场近似)。如图4所示,速率控制因子和选择性控制因子的计算结果表明,CO直接氧化(*→OOs*)和OCOO(OOs*→OV*)路径主导了低温CO氧化活性,H2直接氧化(*→OOs*)和氧迁移路径(OOs*→OV*)是导致高温选择性下降的关键。进一步结合速率贡献度分析,将主导路径涉及的所有基元反应组合为简化PROX网络。与全反应网络求解结果的对比分析表明,简化网络能够实现对CO氧化速率、H2氧化速率和CO2选择性在PROX温度窗口内的近似描述。
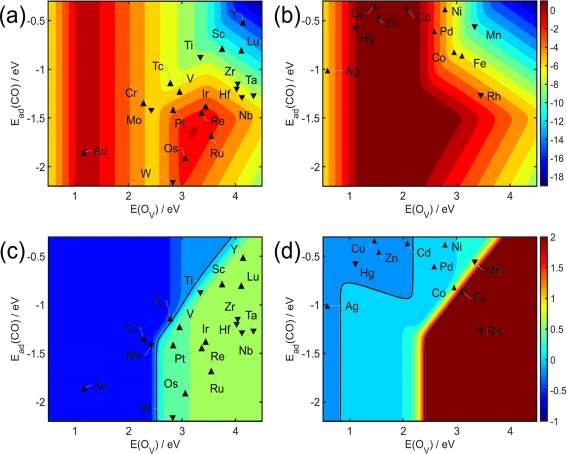
随后,针对所有同质DACs进行简化网络的批量DFT计算,SISSO方法推荐的PROX性能描述符指向解离O2的氧去除能(或氧空位形成能,EOv)、CO和H在金属位点的吸附能,其与CO氧化活性和选择性的关系如图5所示。H吸附能适中(-0.6~0.0eV)时,CO吸附能介于-2.0~-1.5eV、氧空位形成能介于2.6~3.2eV是兼具良好CO氧化活性和CO2选择性的理想范围,且该区间可随H吸附的减弱而进一步扩大。Ir1-Ir1和Ru1-Ru1等贵金属DACs在同质DACs中接近活性火山图的顶端,同时也展现出良好的选择性,与实验报道相符,但PROX性能仍具提升空间。
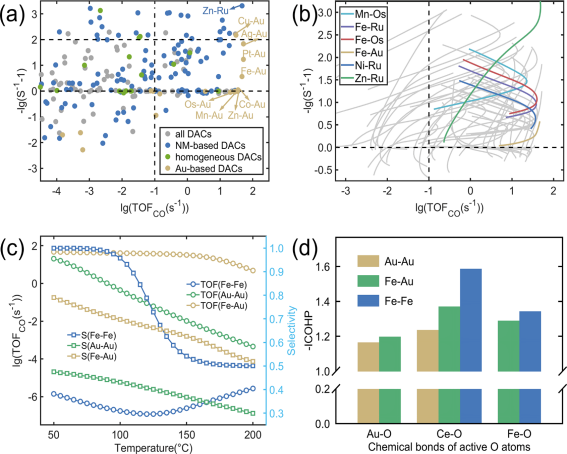
而后,面向所有DACs针对PROX性能描述符进行高通量计算,从而预测其CO氧化活性和CO2选择性,如图6所示。部分金基和贵金属基DACs,如Fe-Au、Cu-Au和Zn-Ru等,展现出优异的低温PROX性能。其中,Fe-Au、Fe-Ru等6种DACs在整个工作温度窗口均保持TOF>0.1s-1且选择性>50%,被认为是DACs/CeO2中最具潜力的PROX催化体系。最后以Fe-Au为例,分别针对Fe-Fe、Au-Au和Fe-Au进行微反应动力学对比验证和电子结构分析。结果表明Fe-Au兼具了Au-Au高活性和Fe-Fe高选择性的优势,这源于Au、Fe对活性氧结合强度以及氢吸附强度的有效协同调控。
综上所述,本项研究基于团队提出的态到态复合方法,面向金属氧化物负载型过渡金属双原子体系的CO PROX过程,开展了第一原理计算结合微反应动力学分析的机器学习辅助高通筛选工作,发现了数种潜在高效的双原子PROX催化剂,为负载型DACs的理性设计和性能调控提供了理论指导准则。